
| |
Manual |
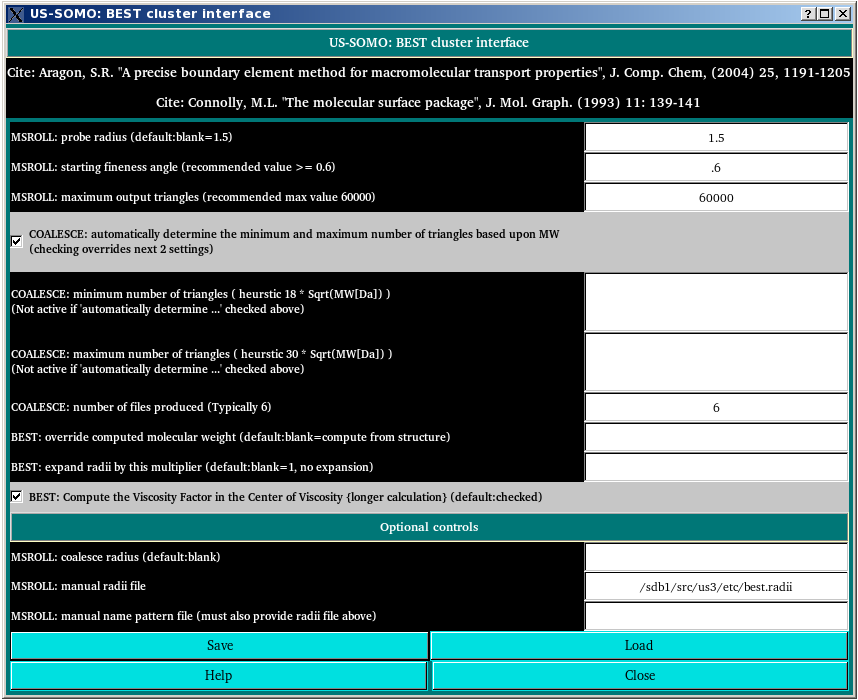
In this module you can set-up all the parameters required to run the computation of the hydrodynamic parameters from an atomic-resolution structure using the boundary elements method BEST [S.R. Aragon, A precise boundary element method for macromolecular transport properties. J. Comp.Chem., 25, 1191-1205 (2004); S.R. Aragon and D.K. Hahn, Precise boundary element computation of proteins transport properties: Diffusion tensors, specific volume and hydration, Biophysical Journal, 91:1591-1603 (2006)] as implemented on a specific supercompute cluster through the US-SOMO interface. BEST requires the ausiliary program MSROLL [M.L. Connolly, The molecular surface package. J. Mol. Graph. 11, 139-141, 1993] to produce an initial triangulation of the molecular surface. Therefore, the MSROLL parameters can be also set here.
In the first field, the MSROLL probe radius can be set (default: 1.5 Å).
The MSROLL starting fineness angle, which controls the fineness of the sampling in the tessellation rendering algorithm, is set next (recommended: ≥ 0.6; default: 0.6).
In the third field, the MSROLL maximum output triangles is set (recommended and default max value: 60,000). In principle, the larger this number, the more accurate will be the representation of the molecular surface. In practice, a too large number of triangles for relatively small structures can lead to singularities, and there are also practical computer memory limits. The default max value of 60,000 triangles was optimized for the supercompute cluster in which the US-SOMO BEST application is run.
The next four options are used for the definition by the BEST module COALESCE of the number of "plates" (triangles) in a structure used to compute the hydrodynamics, and how many such structures should be generated in order to allow for a correct extrapolation to zero plates number.
The checkbox COALESCE: automatically determine the minimum and maximum number of triangles based upon MW uses an automated heuristic approach to determine the lower and upper limits of the triangles used to represent a structure. The formulas used are:
"minimum number of triangles = 18 * sqrt(MW)"
and
"maximum number of triangles = 30 * sqrt(MW)"
with MW in Daltons. (default: checked). If the checkbox is deselected, users can manually enter the minimum and maximum number of triangles values in the COALESCE: minimum number of triangles and COALESCE: maximum number of triangles fields, respectively (advice: between 2,000 and 7,000 triangles).
The number of independent triangulated structures produced by COALESCE is set in the COALESCE: number of files produced field (default: 6; this is a good compromise beween computational speed and a enough points to perform a sound extrapolation).
The next two fields and a checkbox refer to the main BEST program settings:
BEST: override computed molecular weight allows entering a different molecular weight from that computed from the composition, as present in the PDB file. It effects mainly the intrinsic viscosity calculation, since BEST doesn't compute directly the sedimentation coefficient (default: blank, i.e. compute from structure)
BEST: expand radii by this multiplier allows an expansion of the atomic radii stored in the best.radii file that were optimized to account for an uniform layer of hydration (default: blank =1, no expansion).
Checkbox BEST: Compute the Viscosity Factor in the Center of Viscosity (longer calculation) permits a more accurate evaluation of the intrinsic viscosity (default: checked). It should be deselected to speed up the computations when there is no interest in accurately computing the intrinsic viscosity.
By clicking on the Optional controls bar, three more fields related to MSROLL settings become available, as shown in the image above.
MSROLL: coalesce radius. This field defines an optional coalescing stage in MSROLL.
MSROLL: manual radii file. In this field you can change the atomic radii file used by MSROLL (default: best.radii).
MSROLL: manual name pattern file (must also provide radii file above). This file defines the names used in the atomic radii file above and is defined in the MSROLL manual (version 3.9.3 chapter 16). If you are defining your own pattern files, you should consult the MSROLL reference documentation.
Save will save in a file the current set of parameters, which can be retrived through the Load button.
Close: Exits and remembers all changes made to the values in preparation for creating a cluster job package for submission.
After cluster packaging, submission, retrieval and results extraction, a series of files will be produced in the results directory and are described here:
Note: the filename components _f#_##, if present, describes the fineness associated with the file and the final _#####, if present, defines the number of triangles.
| File extension or pattern match | Description |
| .csv | These are the primary results for analysis within the BEST analysis module or can be examined with a standard spreadsheet program |
| .be | The BEST run output |
| .log | The BEST run log file |
| _s.pdb | A copy of the processed PDBs |
| _s-removed.pdb | These will contain any atoms removed from the submitted PDBs before processing |
| .c3p | The MSROLL edges and vertices |
| .c3v | The MSROLL summary information |
| .bead_model | A bead model of the surface produced |
| msr*.stdout | Output of the MSROLL runs |
| msr*.sterr | Error output of the MSROLL runs |
| rcoal*.stdout | Output of the coalesce runs |
| rcoal*.sterr | Error output of the coalesce runs |
| best*.stdout | Output of the BEST runs |
| best*.sterr | Error output of the BEST runs |
| No file extension | Tessellation edges and vertices produced by coalesce |
| errors-# | Possible error messages which caused a run to fail |
| notice-# | Notices about less significant issues during the run |
This document is part of the UltraScan Software Documentation
distribution.
Copyright © notice.
The latest version of this document can always be found at:
Last modified on May 11, 2014